
Home



Directory
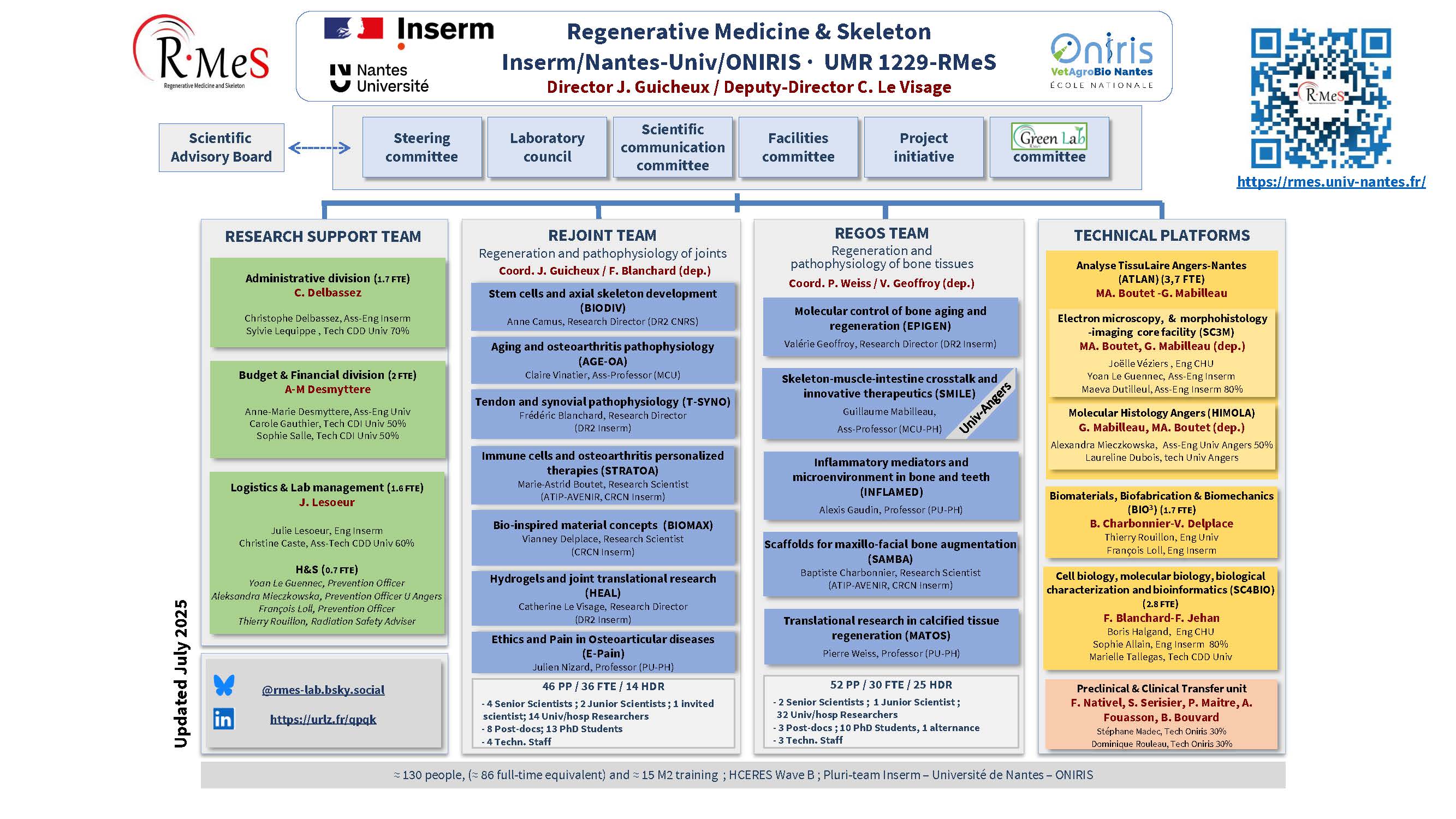


Intranet / wiki
RMeS members only
To log in, please use your institutional credentials
Others contents

Updated on 05 March 2026.